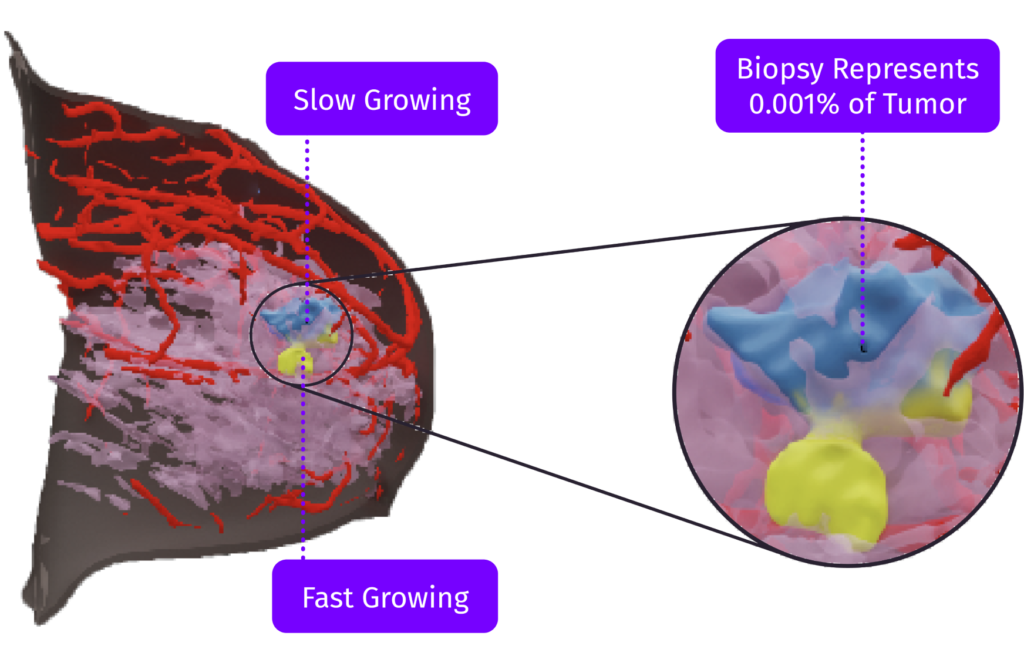
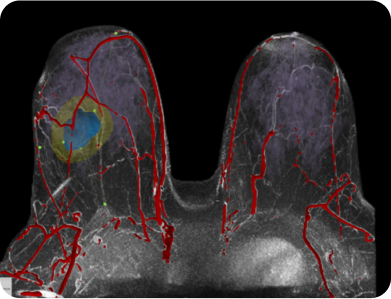
“This (TumorSight Viz & Plan) can really aid in my surgical planning. It can inform the optimal incision placement and those who likely need less or more oncoplastic rearrangements. If we’re doing breast conservation, are we going to need to take the nipple-areola complex or not?"
Alastair Thompson, MD Baylor College of Medicine